V) NMR Spectroscopy and Analysis of NMR Parameters
5. Identification of Frozen Transition States
(Professor Dieter Cremer, Professor Elfi Kraka, and co-workers)
The major goal of Chemistry is to control chemical reactions. This goal can only be fulfilled if one has exact knowledge about the transition states (TSs) of chemical reactions. Unfortunately, detailed information about the properties of TSs is very difficult to obtain by experimental means, which has to do with the fact that a TS is a transient point of the reaction path, at which the reacting molecules do not stay for any period of time needed for measurement. Out of this dilemma, namely a) to need detailed information on TS properties and b) not being able to directly investigate a TS, the idea of frozen TSs was born. If it would be possible to increase the stability of a TS relative to that of reactant and products, a point should be reached at which the TS becomes a minimum on the potential energy surface (PES) while reactants and product occupy transient points on the PES, i.e. the chemical reaction is frozen at the TS. In this situation, the TS geometry, its charge distribution, its dipole moment, its magnetic properties and all other properties become measurable because of the finite life time of a system sitting at a minimum of the PES. Realization of frozen TSs has been a major goal in experimental chemistry because it should lead to the urgently needed knowledge about TS properties. However, realization has met too many obstacles and, accordingly, remained unsuccessful for a long time.
We discovered that various homoaromatic compounds such as the homotropenylium cation represent frozen TSs and that they provide ample information on the properties of TSs. An even more impressive example was found when investigating substituted semibullvalenes since appropriate substitution leads to freezing of the TS of the Cope rearrangement.
For further reference, see:
- 116Homotropenylium Cation: Structure, Stability, and Magnetic Properties, D. Cremer, F. Reichel, and E. Kraka, J. Am. Chem. Soc., 113, 9459 (1991).
- 148Cyclopropyl Homoconjugation, Homoaromaticity and Homoantiaromaticity - Theoretical Aspects and Analysis, D. Cremer, R.F. Childs, and E. Kraka, in "The Chemistry of Functional Groups, The Chemistry of the Cyclopropyl Group", Vol. 2, ed. by Z. Rappoport, John Wiley, New York, 1995, p. 339.
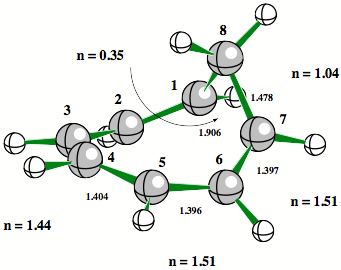
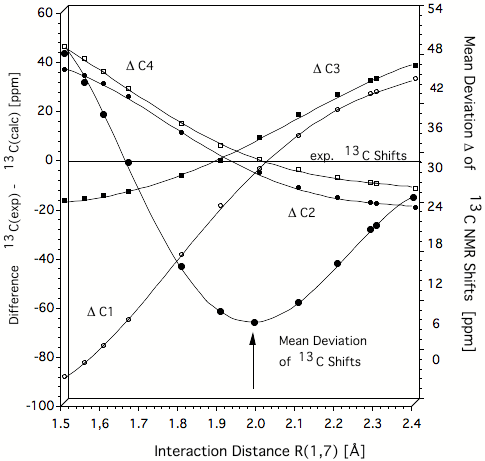
Figure 1. Difference Δ between experimental and calculated IGLO/6-31G(d,p) 13C NMR chemical shifts of the homotropenylium cation as a function of the 1,7 distance. The best agreement between calculated and experimental chemical shift values is obtained at 2 Å. This is thereby the true C1,C7 distance typical of the C..C distance of a breaking/forming bond in the TS. Hence, the homotropenylium cation is a frozen TS where the bicyclic form with short C1C7 bond (1.5 Å) and the open, monocyclic form with long C1C7 distance (2.4 Å) are the reference structures (reactant and product). All values in ppm.
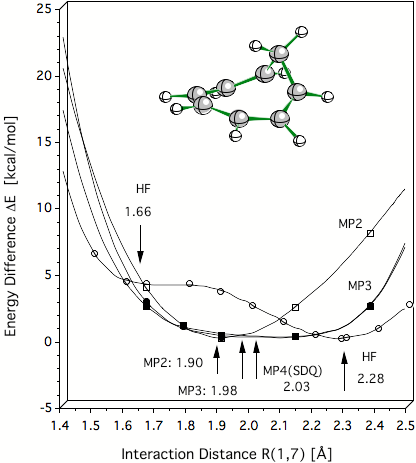
Figure 2. Verification of the C1C7 distance of 2 Å for the homotropenylium cation by ab initio calculations. HF theory is unable to describe the equilibrium of the homotropenylium cation and suggests a double well potential. All MP methods, however, predict a single well potential where the equilibrium distance C1C7 converges to a value of 2 Å (predicted by MP4 and CCSD(T), not shown).
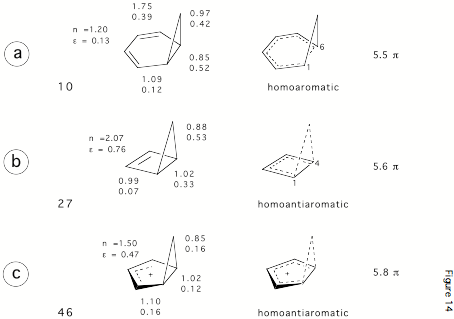
Figure 3. Some bicyclic hydrocarbons with potential homoaromatic or homoantiaromatic character. Bond orders n and p-bond ellipticities e, and the number of p-electrons in the conjugated ring where the 3-membered ring bond is considered to be a p-bond.

Figure 4. Different forms of electron delocalization.
6. Investigation of Magnetic Susceptibilities and Susceptibility Exaltations as an Important Tool for Analyzing Electronic Structure Changes during a Reaction.
(Professor Elfi Kraka, Professor Dieter Cremer)
In past work we could show for the homotropenylium cation and some other aromatic system that an increase in the magnetic susceptibility indicates an increase in electron delocalization, which is in line with Dauben's approach to describe aromaticity with the help of susceptibility exaltations (a renaissance of Dauben's work can be found in the publications of Paul v. R. Schleyer). Our goal was to investigate changes in the magnetic susceptibility along a reaction path. Since we had to substitute correlation corrected ab initio methods more and more by DFT methods because of cost considerations we extended the theory of calculating magnetic susceptibilities to DFT methods. We found (as well as other authors) that the electron density obtained by a DFT method is too small at the position of the nuclei, hence the nuclei are not so strongly screened, the calculated energies of the occupied orbitals are too high, and paramagnetic effects are too strong (relevant for chemical shifts) whereas diamagnetic effects are slightly too weak (relevant for magnetic susceptibilities). Based on this understanding, we included corrections into a DFT method that increase the electron density at the nuclei and, by this, lower orbital energies. According to Schleyer, the susceptibility exaltation is the only reliable electron delocalization criterion and therefore we exploited this criterion in our studies on reaction mechanism (in particular for the pericyclic reactions).
For further reference, see:
- 116Homotropenylium Cation: Structure, Stability, and Magnetic Properties, D. Cremer, F. Reichel, and E. Kraka, J. Am. Chem. Soc., 113, 9459 (1991).
- 148Cyclopropyl Homoconjugation, Homoaromaticity and Homoantiaromaticity - Theoretical Aspects and Analysis, D. Cremer, R.F. Childs, and E. Kraka, in "The Chemistry of Functional Groups, The Chemistry of the Cyclopropyl Group", Vol. 2, ed. by Z. Rappoport, John Wiley, New York, 1995, p. 339.
7. Direct Detection of Hydrogen Bonds in Biopolymers Using NMR Spin-Spin Coupling Constants
In biochemistry, hydrogen bonds are responsible for the selectivity of base pairing in nucleic acids, which is the basis for the preservation and propagation of genetic information; they are also important for the three-dimensional folding of proteins because the most prominent structural features of proteins (e.g. helical and a-sheet regions) are defined and stabilized by the unique pattern of hydrogen bonds. Finally, intermolecular hydrogen bonds contribute to the affinity and selectivity of molecular recognition ranging from simple host-guest systems to multi-component complexes of proteins, nucleic acids, etc.
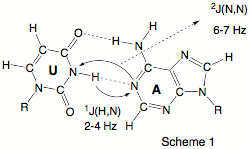
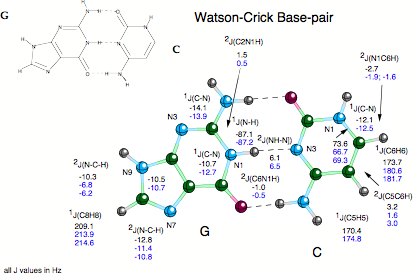
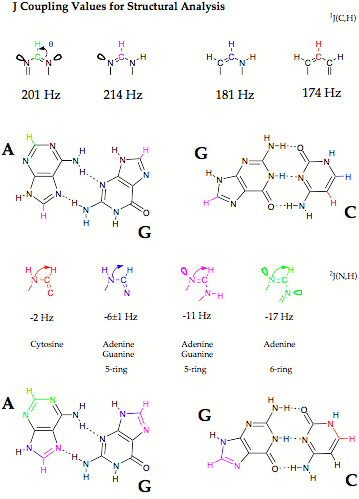
Despite their importance, commonly used techniques such as X-ray diffraction or NMR spectroscopy provide only indirect evidence of the existence of hydrogen bonds, e.g. by the location of possible H-donor (D) and H-acceptor (A) atoms (X-ray). In 1998, Grzesiek (J. Am. Chem. Soc. 1998, 120, 8293) reported the observation of scalar spin-spin coupling constants (SSCCs) across the hydrogen bonds in the Watson - Crick base pairs of a 15N-labeled 69 nucleotide RNA (see Scheme 1a). Grzesiek’s measurements were confirmed in the last year by other authors and are now considered as the first direct information on H-bonding in biochemically important molecules thus triggering a manifold of NMR projects in biochemistry.
In calculations on some small H bonded dimers we demostrated that NMR SSCCs across H bonds can be accurately described with the CPDFT method programmed by us.
- Calculation and analysis of across H bond couplings in various H-bonded complexes. For a number of small H-bonded complexes with N, F, O as H donor or acceptor atoms, SSCC values were calculated and analyzed to obtain a basic understanding of the nature of the electronic coupling mechanism. Beside SSCCs, we also calculated NMR chemical shifts with our DFT-IGLO method to test whether H bonding can also be characterized with NMR chemical shifts (e.g. covalent/electrostatic nature; the SSCC mechanism requires some covalent nature; shielding or deshielding of the proton shift reflects the electrostatic nature of H bonding). We used the topological analysis of the electron density distribution and the adiabatic mode concept developed by us to obtain a detailed description of H-bonding.
- Calculation of across H bond coupling constants for Watson-Crick base pairs. There is a large number of possible base pairs made up from adenine, guanine, thymine (uracil), and cytosine as e.g. Watson-Crick, Hoogsteen, Reverse Watson-Crick pairs, etc. (quantum chemical information on more than 30 base pairs can be found in the literature). We investigated those base pairs, for which NMR investigations have been done already. The investigation of across H bond coupling constants was combined with the calculation of other SSCCs and NMR chemical shifts to get a detailed description of the base pairs.
- Dependence of across H bond coupling constants on geometrical features. We determined how the location of H donor and H acceptor influences the magnitude of across H bonding coupling where distance and angular variation was first carried out for model complexes and then for some of the most interesting base pairs.