III) Reaction Mechanism and Reaction Dynamics
B) Special Reaction Systems
1. Investigation of the C4H4 Potential Energy Surface
(Project leaders: Professor D. Cremer and Professor E. Kraka, in collaboration with Professor T. Zwier, Purdue University)
Certain planets in our sun system possess an atmosphere that is rich on hydrocarbons. For example, Saturn’s moon Titan is known to possess benzene and other larger hydrocarbons in its atmosphere, which are probably formed by photochemical reactions of C4H4 molecules. At Purdue University, Titan’s atmosphere was simulated and exposed to Laser light flashes and the resulting chemical compounds analyzed. At Pacific we calculated all possible reactions of C4H4 molecules so that we were able to explain the experimental observations and to describe the way benzene is formed in Titan’s atmosphere.
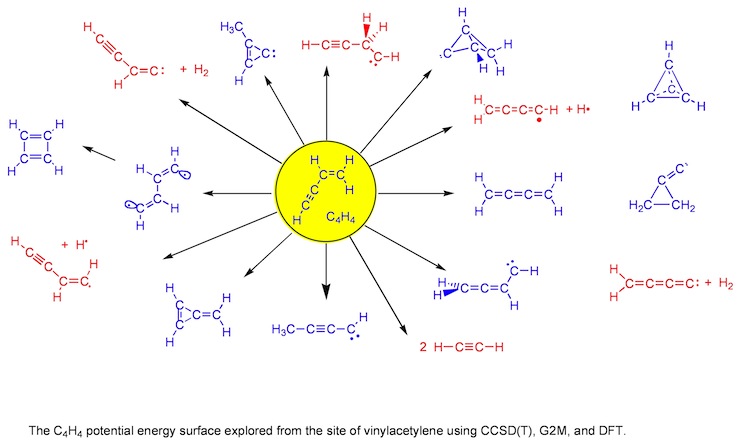
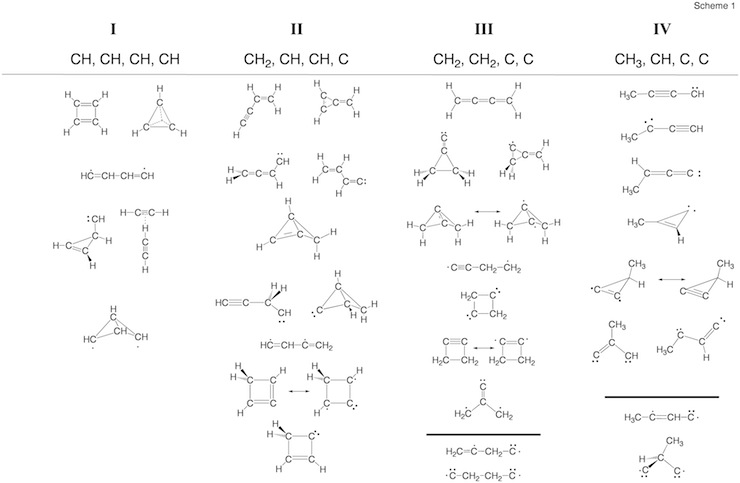
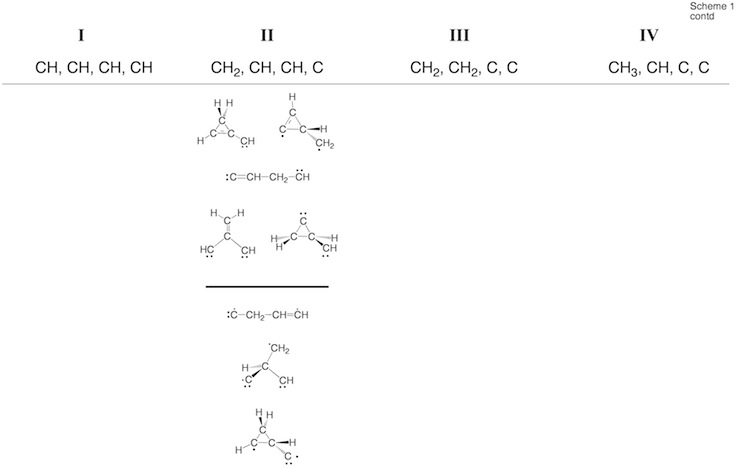
Figure 1. Systematic search for chemically reasonable structures to be found on the PES of C4H4.
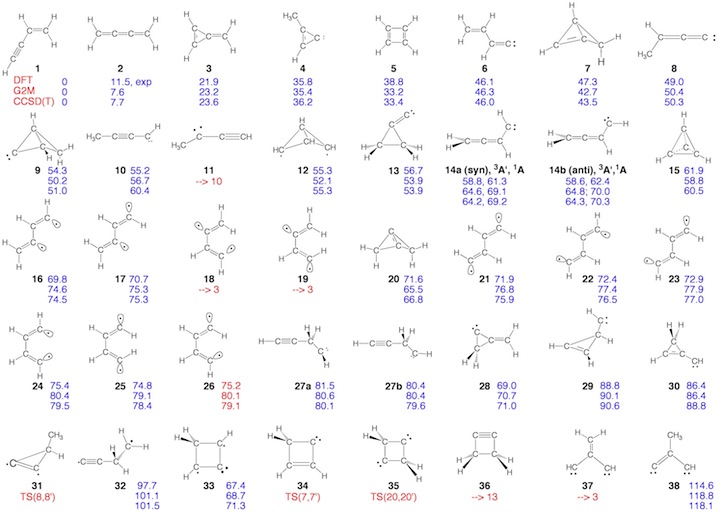
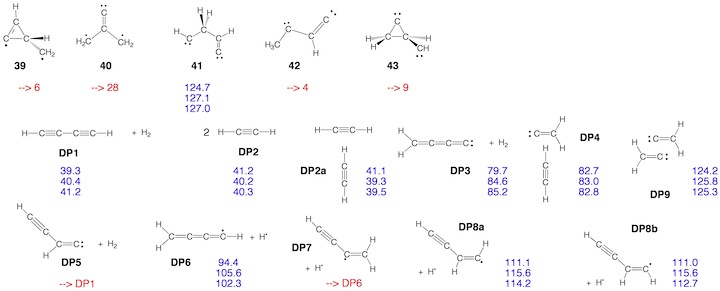
Scheme 2. Minimum C4H4 structures 1 – 43 and dissociation products DP1 – DP9 located at the on the C4H4 PES. For each structure the relative enthalpy value DDH(298) (reference is 1) calculated at the DFT (top entry), G2M (middle entry), and CCSD(T) level of theory (bottom entry) is given. Some structures were identified as TSs, others rearranged to more stable structures. For 2, the DFT level (2.9 kcal/mol) is far below the experimental value of 11.5 kcal/mol so that the latter is given. For carbene 14, values for both the T ground state and the S excited state are listed.
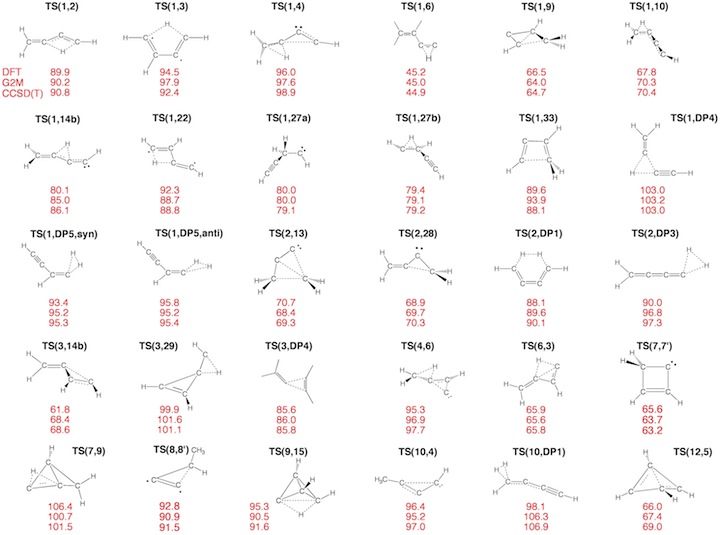
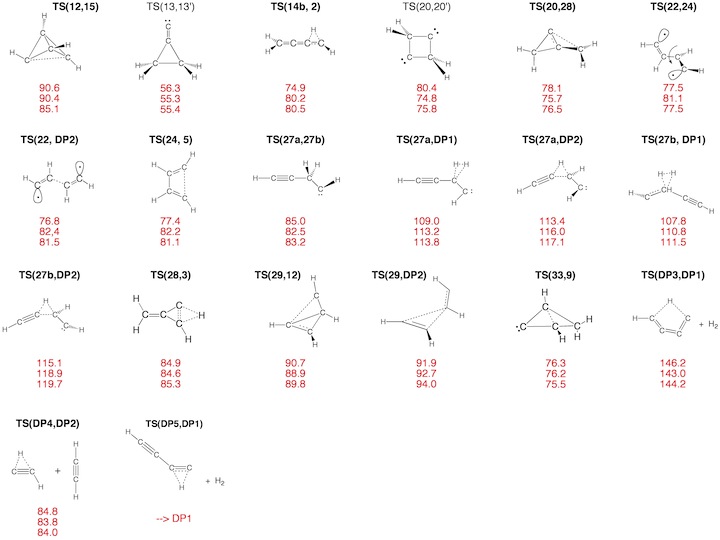
Figure 3. Transition State (TS) structures of reactions R1-R30 taking place on the C4H4 PES. For each structure the relative enthalpy value DDH(298) (reference is 1) calculated at the DFT (top entry), G2M (middle entry), and CCSD(T) level of theory (bottom entry) is given.
2. Characterization of Allenylcarbene
(Project leaders: Dr. E. Kraka and Dr. D. Cremer)
Allenylcarbene has been suggested as an intermediate on the C4H4 potential energy surface. First spectroscopic results for this compound have been obtained by matrix isolation spectroscopy close to the absolute zero point in temperature where however the interpretation of these results is a matter of controversy. The high-level quantum chemical calculations are aimed at providing a reliable description of the three lowest electronic states for different conformations of allenylcarbene and to clarify whether the compound was really found in experiment.
3. Description of Chemical Reactions with Coupled Cluster Methods
(Professor Elfi Kraka and co-workers)
High-accuracy data for potential energy surfaces (PES) can be obtained from MRD-CI, CCSDT, CASPT2, and similar methods. These methods require a considerable amount of computer time, but this is also true for other high-accuracy methods. The real problem with methods such as CASPT2, etc. is that analytical energy derivatives needed for the calculation of stationary points on the PES, reaction path searches, and curvature evaluations at the stationary points (i.e., calculation of vibrational frequencies) are not generally available. Because of this, one concentrates on single- or few-determinant methods for which analytical energy derivatives are available. Most promising in this respect are the Coupled Cluster (CC) methods with single (S), double (D) and triple (T) excitations. CCSD(T) and CCSDT have proven to cover both dynamic correlation effects and substantial multi-reference effects and, therefore, to lead to an accuracy being sufficient for PES investigations.
At the first stage of the project, we calculated stationary points along the reaction path of various reaction systems (combustion reactions, valence tautomeric equilibria, pericyclic reactions, etc.). We investigated which basis sets are needed to describe a given problem with an accuracy of 1 kcal/mol or better at the CC level of theory. In this connection, quadruple and pentuple zeta basis sets augmented with several sets of d, f, and g polarization functions were used. In addition, vibrational, temperature, tunneling, etc. corrections were calculated.
At the second stage of the project, investigations with the reaction path hamiltonian were carried out to learn how energy is transferred along the reaction path and how energy is distributed among the vibrational modes of reactants and products, which is important for mode selective enhancement of reaction rates by Laser pumping.
For further reference, see:
- 128Accurate Couple Cluster Reaction Enthalpies and Activation Energies for X + H2 → XH + H (X = F, OH, NH2, and CH3), E. Kraka, J. Gauss, and D. Cremer, J. Chem. Phys., 99, 5306 (1993).
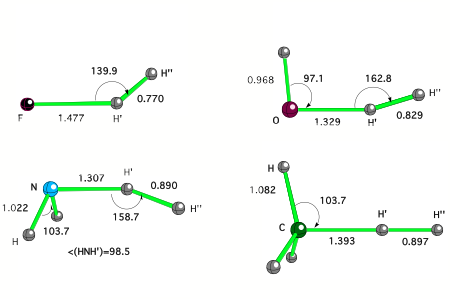
Figure 1. CCSD(T) transition states of some typical XHn + H2 reactions.
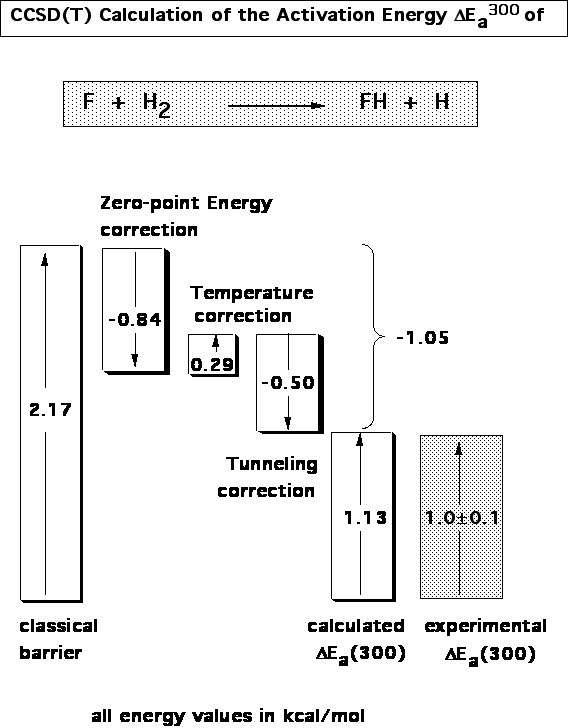
Figure 2. Decomposition of the activation energy of the reaction F + H2 → FH + H according to CCSD(T) calculations.