V) NMR Spectroscopy and Analysis of NMR Parameters
8. Investigation of a DNA Hairpin Molecule: Systematic Investigation of DNA base pairs.
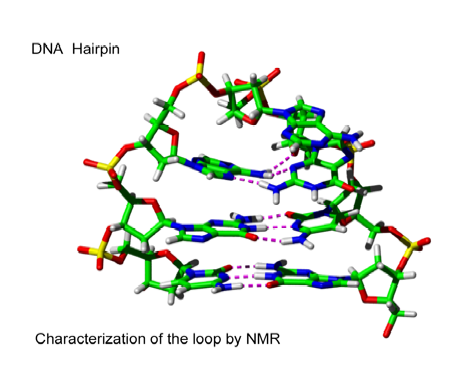
In a joined project with Professor P. Hobza (Heyrovsky Institute, University of Prague) and Professor V. Sklenar (Masaryk University, Brno, Czech Republic) we investigated the structure of a DNA hairpin molecule of the sequence d(GCGAAGC) = d(G1C2G3A4A5G6C7) in a two-pronged approach combining NMR measurements and quantum chemical calculations. The DNA hairpin molecule was labeled by 15N, and therefore we focused on 13C, 15N-coupling constants. Also, coupling constants across the H-bonds of the DNA hairpin molecule were investigated to identify and characterize the interactions between the various bases of the molecule. This concerned two Watson-Crick GC base-pairs G1C7 and C2G6, a mismatched base-pair G3A5, and an unpaired adenine base A4. Different types of H-bonding had to be identified and characterized via the J values across the H-bonds.
The most relevant base pairs of the full DNA hairpin molecule were modeled in the calculations (base-pair plus ribose rest). The influence of the environment was considered by carrying out solution phase calculations. Non-specified salvation by water was considered by a continuum model of the appropriate dielectric constant whereas specific solvation was modeled by investigation of individual water complexes.
9. Investigation of the Protein Ubiquitin: Systematic Investigation of Polypeptides and Proteins.
The secondary structure of proteins is largely determined by the formation of specific H-bonds, which can be described by the NMR spin-spin coupling constants across the N-H...O=C fragment. Experimentalists have succeeded in routinely determining the 3hJ(N,C) constants of polypeptides and proteins. For the protein ubiquitin (76 residues) that plays in almost all eukaryotic cells an important role by marking other proteins for degradation and for which a X-ray diffraction structure is available [2], nearly all interresidue 3hJ(N,C) constants across the H-bonds could be reliably measured. Hence, ubiquitin represents an excellent example for a protein, for which the dependence of 3hJ(N,C) on the geometry of the H-bond can be investigated. Our investigation was based on the following strategy:
- We selected 3- to 4-residue-fragments of ubiquitin so that the vicinity of a H-bond could be reliably modeled. Side-chains were replaced by H atoms and the geometry of the fragment was partly reoptimized. For the model structure obtained in this way, we calculated all J-values across the H-bond and compared calculated J constants with the available experimental values.
- In a parallel investigation, we determined nJ constants across the H-bond of the formamide dimer, which was used to model the H-bond in the N-H...O=C fragment of a polypeptide or protein. By changing the geometry of the N-H...O=C fragment systematically, the dependence of the nJ constants on key geometrical parameters was determined and a nJ-hypersurface derived.
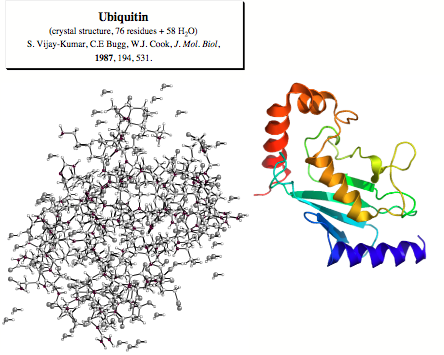
Figure 1. Crystal structure of ubiquitin (76 residues + 58 H2O molecules, see Ref. 2).
Figure 1. X-ray structure of protein ubiquitin.
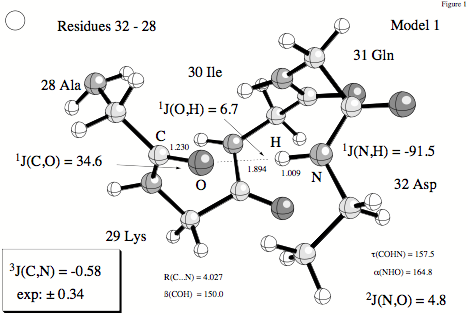
Figure 2. H-bonding between residues 32 (donor) and 28 (acceptor) of the protein ubiquitin. Distances in Å, angles in degrees, and spin-spin coupling constants in Hz.
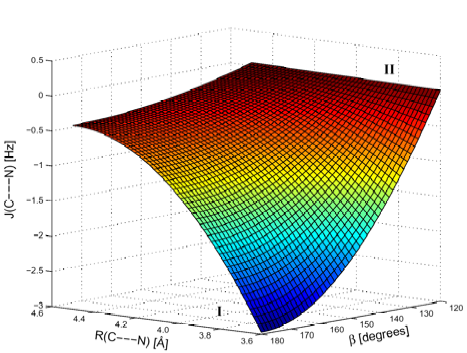
Figure 3. Perspective drawing of the SSCC 3hJ(N.... C') calculated as a function of the distance R(N....C') and the angle ß. Region I and region II indicate geometrical situations, in which spin-spin coupling mechanism I (domination by electric field effect) and II (delocalization of O lone pair) are important.
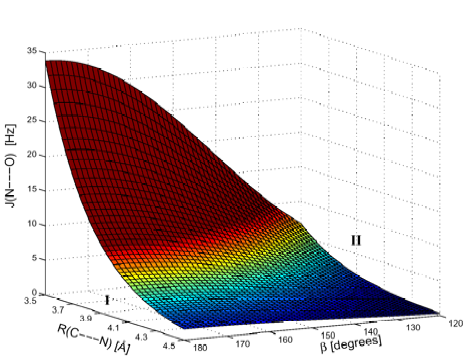
Figure 4. Perspective drawing of the SSCC 2hJ(N.... O) calculated as a function of the distance R(N....C') and the angle ß. Region I and region II indicate geometrical situations, in which spin-spin coupling mechanism I and II are important. Compare with Figure 3.
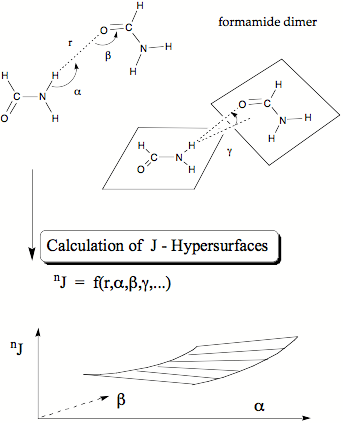
Figure 5. Investigation of the formamide dimer as a suitable model for H-bonding across the N-H..O=C fragment in proteins and polypeptides. By variation of the geometry of the formamide dimer the coupling constants nJ across the H-bond can be expressed as functions of key geometrical features thus leading to a nJ-hypersurface.
10. Molecular Dynamics Calculations to Determine Spin-Spin Coupling Constants Directly Comparable with Experimental Values.
In our recent investigation of the spin-spin coupling across the H bonds of the protein ubiquitin, we found the necessity of using a correct description of the spin-spin coupling mechanism, which previously was just described by its Fermi contact (FC) term. By including also spin dipole (SD), diamagnetic spin orbit (DSO) and paramagnetic spin orbit (PSO) term, the reliability of the calculated SSCCs across the H-bonds was considerably improved and their structural dependence determined. For the direct comparison of measured SSCCs the inclusion of vibrational effects and conformational averaging is necessary. Also it has to be considered that measurements are done in aqueous solution. Therefore, we will use molecular dynamics (MD) simulations in aqueous solution to consider the influence of both vibrational, conformational, and solvation effects. Besides standard MD calculations utilizing the Amber force field, we will also employ Car-Parinello simulations. A generalized Born solvation model will be applied with a modified set of Bondi radii and screening parameters to be adapted from the Tinker programs. Recent simulations of proteins have demonstrated that it is possible to obtain ensemble averages compatible with a particular microscopic state from the trajectory of a single molecule. The details of the calculations have been tested (energy minimization, heating up in the heat bath, slow removal of the heat bath, 500-ps MD run at constant energy dynamics using a 1-fs time-step; saving of conformational geometries every 1 ps, etc.).
We will first investigate ubiquitin to see what changes in the SSCCs are caused by vibrational and conformational movements. In the calculations of the SSCC across the H-bonds we will use the same strategy as described previously by us. We will apply the procedure also to other targets such as an immunoglobulin binding domain of streptoccal protein G, for which experimental SSCCs across H-bonds and a high-resolution X-ray diffraction structure are available. Goal of this work is to use experimental SSCC in connection with the simulated values to point out differences between the crystalline and the solution-phase geometries in the vicinity of the H-bond bridges. Another goal is to predict SSCCs across H-bonds that are difficult to measure by experimental means.
11. Investigation of Through-space Coupling in F-containing Compounds
The physical properties of fluorine (small size, spin of 19F is 1/2, 100\% natural abundance of 19F) coupled with its large gyromagnetic ratio, makes long range NMR spin-spin coupling constants (SSCCs) nJ(19F,19F) = nJ(F,F) important parameters for studying the structure of polycyclic aliphatic carbons, steroids, and aromatic molecules. Indeed, SSCCs nJ(F,F) with large n have for some time already been used as important probes in biochemical investigations. However, the mechanism by which the spin information is transferred from one fluorine atom to the other via a long chain of bonds (through-bond) or alternatively via a direct through-space route is not yet fully understood.
For coupling fluorine nuclei that are proximate to one another (i.e. within the van der Waals distance of 2.94 Å) the transfer of spin information is generally thought to occur through the lone pairs (lp) of the F atoms. However, several cases exist where sizable couplings occur between F nuclei that have a separation larger than 2.94 Å. These long-range couplings have generally been explained by a through-bond mechanism - either via a hydrogen bond or a p-electron system. More recently, the pure through-space limit has been extended to the outer limit of the van der Waals distance, where 398J(F,F) = 17 Hz detected within a dihydrofolate reductase-NADPH-MTX complex, was determined to be transmitted purely through-space.
It is the general understanding that the Fermi contact (FC) coupling mechanism dominates F,F coupling, but it is still not well understood, which orbitals are responsible for through-space coupling in which way, and whether this is exclusively a FC coupling mechanism or whether the non-contact interactions (PSO, DSO, SD) also contribute. Our previous calculations reveal that the SD coupling mechanism can be much more important than the FC mechanism under certain circumstances. The FC coupling mechanism requires that there is spin density at the coupling nuclei, which cannot be expected if p-orbitals are involved. However, the latter can play a passive role and enhance in this way spin-spin coupling. A quantitative assessment of the contribution of the p- or s-lp-orbitals of F is essential for an understanding of the F,F coupling mechanism.
We investigated fluorine-substituted organic compounds such as 1,8-difluoronaphthalene, o-F-benzamides, F-benzoximes, F-benzaldehydes as well as difluoro-substituted proteins, for which long-range F,F or F,N-coupling constants have been measured. We used in this connection the J-OC-PSP method to determine the electronic structure information provided by measured NMR SSCCs between F or between F and N nuclei.
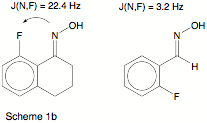
It has been speculated for some time that non-covalent interactions can also lead to SSCCs. Mallory et al. (see, e.g., J. Org. Chem. 57, 366, 1992) measured intramolecular through-space N-F couplings (9-43 Hz) which were explained by a lone-pair orbital overlap theory. If this theory would be correct, it should be possible to detect intramolecular non-covalent interactions with the help of NMR spectroscopy. - The theory of Mallory was checked by calculating SSCCs for fluorinated oximes (see Scheme 1b) and analyzing their origin.